
シアル酸の科学
シアル酸 ―現代新薬の創製まで―
第1章 シアル酸の化学
1 シアル酸の歴史1)
シアル酸は生物発生の歴史と共に存在したものであるが、その存在が明確になったのは、たかだか60年位前のことである。この物質は、酸性アミノ糖で生体の糖脂質や糖蛋白質などからえられたが、運悪く、丁度、第2次世界大戦のさなかであったために、各国研究者のコミュニケ一ションが悪く、必ずしも順調な滑り出しではなかった。
シアル酸の発見は独立した2つの研究グループによって行われた。すなわち、クレンク(E.Klenk)2)の糖脂質の研究とブリックス(G.Blix)3)の糖蛋白質の研究である。クレンクの1935年から1939年にわたる研究で、Tay-Sacks病やNiemann-Pick病(先天性代謝異常症)患者の脳から、“ビアル(Bial)試薬”で紫色に呈色する物質が得られた。ビアル試薬とは、1927年、Walz, Leven, Landsteinerらによって、ウマの腎臓やヒ臓、ウシの腎臓や脳のセレブロシド部分の発色に用いられたものの一つで、オルシノールと3価の鉄イオンを濃塩酸に溶かしたものである(図-7)。普通の糖鎖をもつ脂質ではこの試薬と加熱しても緑色を呈するのみである。
1938年、ブリックスは牛の脳に、この呈色反応陽性の糖脂質があることを発見した。3)更に、1939年、クレンクは痴呆症患者のセレブロシドに強い酸性の脂質が含まれていることを発見した。1941年、これをメタノール中、5%の塩酸で105℃、3時間処理してアミノ基とカルボキシル基をもつ結晶性物質で 、ビアル試薬によって紫色を呈する物質を得て、これに“ノイラミン酸”と名付け、その原料となった糖脂質を“ガングリオシド”と名付けた。2)ノイロ(Neuro-)とは神経の意味である。今日、ノイローゼ(Neurose)は立派な日本語になっている。
1年後、クレンクはこのノイラミン酸はメトキシノイラミン酸であることを確定すると共に、ビアル試薬はこのメトキシノイラミン酸と定量的に発色することを明らかにした。4)この試薬はその後のシアル酸研究の有用な武器となった。

一方、ブリックスは1936年、牛の顎下腺ムチンを水で煮沸し、結晶性物質を得て“Carbohydrate I”と名付けた。この物質はモノカルボン酸で、パラジメチルアミノベンズアルデヒドの塩酸溶液と加熱すると、紫に呈色した。これはエールリッヒ(Ehrlich)反応と呼ばれるものである(図-7)。この酸性物質は希鉱酸と加熱すると、容易に分解してフミンを生成した。更に、2個のアセチル基をもち、その1個は N-アセチル基であることを推論した。2年後、クレンクの方法に従って、ブリックスは人の脳の糖脂質も同様にエールリッヒ反応が陽性であること、希鉱酸と加熱すると同様にフミンを生成すること、ビアルの試薬で紫色を呈することを発見した。5)
このように、シアル酸研究の黎明期は過ぎていったのであるが、クレンクの“ノイラミン酸”とブリックスの“Carbohydrate I”の関係が明かになるのはまだ20年も後のことである。そうこうするうちに、1942年、オーストラリアのファーストはインフルエンザウイルスが赤血球を凝集することを発見した。6)この現象は赤血球の表面にレセプターがあって、これにウイルスが吸着すると考え、この現象がウイルス感染の第一歩と考えた。この凝集反応にムチンを加えて行うと阻止されることから、ムチンは赤血球と同じような働きがあると考えた。6)
クレンクはこの論文からノイラミン酸がレセプターであると考えて、1952年、顎下腺ムチンとウイルスを混合して透析し、ノイラミン酸を結晶として分離したのであるが、この辺で、シアル酸研究会会長、学士院会員山川民夫先生の業績に触れておかなければならない。1950年と言えば、終戦後、まだ日の浅い頃であるが、山川はウマの赤血球膜からビアルの試薬で赤紫色を呈する糖脂質を得て、これを“ヘマトシド”と名付けた。更にこれをメタノリシスして“ヘマタミン酸”を得た。この物質はビアルやエールリッヒの試薬に陽性で、山川は後年、ヒト赤血球と比較検討して、異なるノイラミン酸(シアル酸)であることを見いだした。1)
ブリックスは1936年、牛から取り出した酸性の未知物質(Carbohydrate I)を1952年、シアル酸と命名し、研究を進めた結果、1956年には、ウシのシアル酸は、N-及びO-ジアセチル誘導体、ブタのそれはN-グリコリル誘導体、ヒツジではN-アセチル誘導体で、ウマはジアセチル誘導体であると報告した。1956年、山川はヘマトシドのノイラミン酸がグリコリル誘導体(18)で、ヒトの赤血球から得られたものはすべてアセチル誘導体であることを明らかにした。その頃、ウシの初乳からラクタミン酸、母乳からジャイナミン酸、ウマの血清からゼロラクタミン酸などが分離されたが、いずれも同一のものであることが分かった。7)
1957年、ブリックス、ゴットシャーク、クレンクは共同で論文を発表し、基本化合物のアミノ九炭糖酸をノイラミン酸(neuraminic acid)と呼び、そのアシル誘導体をシアル酸(sialic acid)と総称することを提案した。8)詳細はクレンク教授の弟子のフェラードが、日独シアル酸シンポジウムで講演した、“The Early History of Saialic Acids”に詳しい。9)
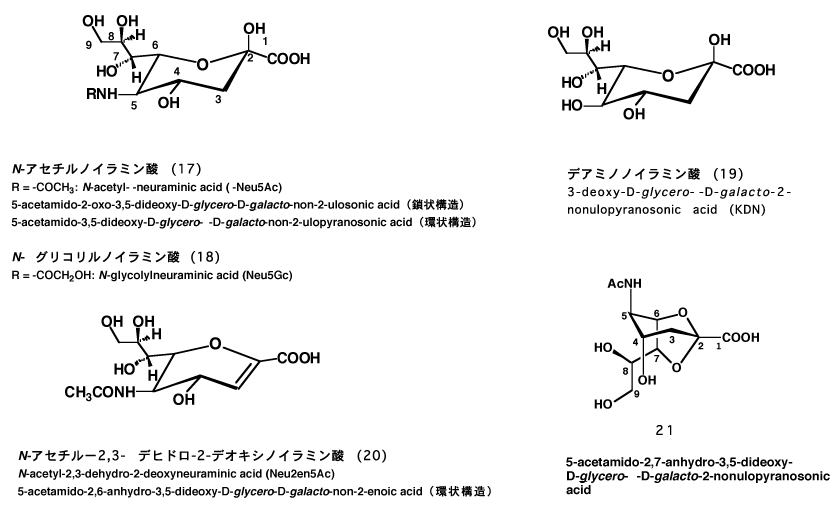
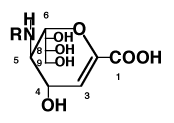
しかし、今日では、ノイラミン酸のアシル誘導体(17、18)のみならず、その他の各種誘導体のほか、アミノ基をもたないデアミノノイラミン酸(19;KDN)をはじめ、デヒドロ誘導体(20)やアンヒドロ誘導体(21)などもシアル酸と呼ぶことを提案している。10)(図-8)
2 N-アセチルノイラミン酸11)
N-アセチルノイラミン酸は従来、生化学の研究にもっぱら用いられていたから、試薬としても、それほど大量に使用されるものではなかった。そのせいもあって、試薬リストから見た定価は大変高価である。例えば、シグマ1996年版によると、合成品(マンノサミンとピルビン酸から酵素を用いる;純度95%)、373.10ドル/g;大腸菌を原料とするもの(純度98%)、405.90ドル/g;羊の顎下腺ムチンを原料とするもの(純度98%)、167.3ドル/250mgである。最近のリストには無くなっているが、かつて、原料が“ヒト”というものがあって、225ドル/gであった。当時、フランスの男性患者にシアル酸代謝異常があって、尿中に1日5〜7gのN-アセチルノイラミン酸を排泄していた。
現在ではグルコサミンを原料とした酵素合成法や大腸菌由来のN-アセチルノイラミン酸を安価に入手できるが、著者らがシアル酸研究を開始した昭和50年頃は大変高価であった。今でも試薬リストを見て頂けば分かるように合成原料としては使えない値段である。そこで横浜の中華街へ行き、食材として売られている破砕された燕窩を購入して原料とした。当初はたしか、一斤6千円だったものが、10箱、20箱と買ううちに瞬く間に品薄となり、一斤十数万円にもなったことを記憶している。
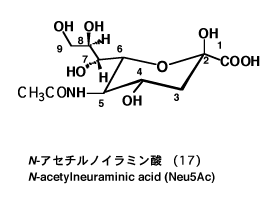
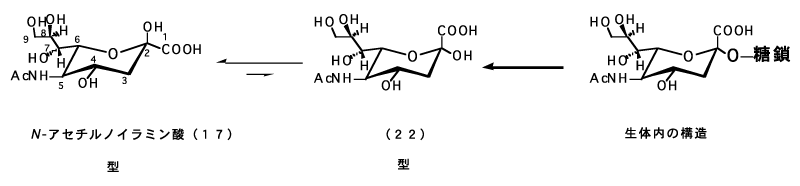
N-アセチルノイラミン酸(17)の構造は図-9に示してあるように、面白い構造の九炭糖であって、結晶ではβ型をとる。(図-10)生体内では、α型(22)をしている。例えば、燕窩を希硫酸で加水分解すると、糖が外れてβ型になる。しかし、純粋なN-アセチルノイラミン酸(17)を水に溶かして核磁気共鳴スペクトルを測定すると、5〜8%のα型を観測することが出来る。N-アセチルノイラミン酸の結晶は再結晶溶媒によって異なる形状を示すが、立体配置はいずれもβ型で同一である。(表-10)
| 再結晶溶媒 | Fine needles (A) | Prisms (B) |
| (C11H19NO8; mp 185-187℃) | (C11H19NO8.2H2O; mp 146-148℃) | |
| Dioxane-water | 1 | 11 |
| Isopropyl alcohol-water | 1 | 1 |
| Acetic acid-water | 1 | 0 |
この結晶構造をX線解析法とCD分析法を用いてれを明らかにしたのはあまり昔のことではないのである。12)古い教科書を見ると、N-アセチルノイラミン酸の立体配置をα型で示してあるものが多い。留意すべきである
3 N-グリコリルノイラミン酸(N-グリコロイルノイラミン酸)
N-グリコリルノイラミン酸(18;Neu5Gc)は哺乳類ではウマ、ブタ、ラット、イヌの一部などに見られるシアル酸である。11,13)ウニなどもこのシアル酸を含むので、ウニの殻を集めて抽出してみたこともあったが、微量しか得られなかった。最近では、ヒトのガン組織や白内障患者の眼液から、微量のN-グリコリルノイラミン酸が発見されているから、生化学的にも重要性を増すものと考えられる。
N-アセチルノイラミン酸(17)を酸で脱アセチル化すると、収率がよくない。大部分が分解するようだ。そこで、図-11に示す方法で、メタンスルホン酸を用いて脱アセチル化して、良好な収率で目的化合物を合成した。14)この反応では、我々の研究室で開発した、N,N'-ジスクシンイミジルカルボナート(23;DSC)を用いた。15)この試薬は、室温で緩和な条件下、簡単に活性エステル(24)を合成出来て、しかもホスゲンとちがって、毒性のない便利なものである。今でも、アルドリッチやフルカの試薬カタログに掲載されている。15)DSCを用いれば、容易にベンジルグリコシドを合成出来るから、高収率でN-グリコリルノイラミン酸(18)を得ることが出来る。
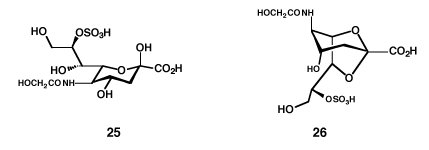
この反応を応用して、ウニの卵巣のグリコリピドの部分構造、N-グリコリル-8-スルホノイラミン酸(25)を合成した。16)また、チオグリコシドを用いて、2,7-アンヒドロ-3,5-ジデオキシ-N-グリコリル-8-スルホノイラミン酸(26)を合成した。(図-11)スルホノイラミン酸誘導体には、エイズ治療予防薬開発の期待があり、現在、中国で研究を続けている。
4 デアミノノイラミン酸(3-デオキシ-D-グリセロ-D-ガラクト-2-ノヌロン酸;KDN)
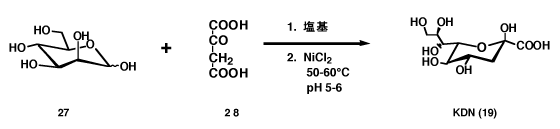
KDN(19)は井上らによって、はじめ、ニジマスの未受精卵から発見された新顔のシアル酸である。17)東北から北海道にかけて、人工受精で盛んに増殖している白鮭にも当然存在して、受精の際に分離すると考えて受精卵のろ液を検討したが、単離精製が困難でKDNの原料としては使用出来なかった。18)KDNはわれわれが開発した合成法で入手可能となった。D-マンノース(27)とオキザル酢酸(28)との縮合反応で、収量よく得られることを明らかにした。19)(図-12)
| 縮合条件 | NiCl2 | Yield(%) | 参 考 | |
| KDN | ||||
| 11) | pH 10 (aq.NaOH) |
10 mol%3) | 11 | 縮合反応の液性が収率に影響する |
| 22) | pH 10 (aq.NaOH-NiCl2) |
10 mol% | 13 | 縮合と同時に脱炭酸すると収率が悪い |
| 31) | pH 11 (aq.NaOH) |
1 mol% | 69 | pHは11がよい |
| 41) | pH 11 (aq.NaOH-Na2CO3) |
1 mol% | 70 | 縮合反応終了後に脱炭酸すると収率がよい |
| 54) | pH 11 (aq.NaOH) |
1 mol% | 66 (KDO) |
KDO合成はこの方法がよい |
1) 反応終了後 NiCl2を加えて脱炭酸
2) 反応混液に NiCl2 を加える
3) オキザル酢酸に対するモル比
4) D-アラビノースを出発原料とする
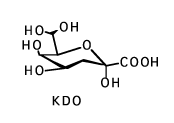
この反応でD-マンノースの代わりにD-アラビノースを用いれば、2-ケト-3-デオキシオクトン酸(KDO;29)が収率よく合成できて応用範囲が広いものである。KDNを合成するこの反応は世界中で利用されている。
KDN(19)はN-アセチルノイラミン酸(17)と異なって、エステル化反応などに複雑な経路を示すから、誘導体の合成に際しては充分注意する必要がある。20)
デアミノノイラミン酸ははじめマス科魚類で研究が進んだが、ヒトも魚類から進化したものであるから、発生段階やガン化組織などに発現して初期ガンの検出に利用できると考え、ドイツの研究者にサンプルを提供したことがある。最近になって、KDNは生物界に広く分布して、バクテリア莢膜、魚類、両棲類、哺乳類の糖タンパク質や糖脂質の糖鎖に存在が確認されるようになった。特に、発生段階特異的、臓器特異的に発現すること、ある種のガン細胞に出現することなどが分かってきた。著者らの合成したKDN誘導体がこの研究では利用されてKDNの生合成経路も明らかにされたが、細胞内のD-マンノースの濃度がKDN濃度と比例することは合成の場合と同じで面白い。今後、更にこの部門の研究は進むと考えられる。21)
5 2,3-デヒドロノイラミン酸
2,3-デヒドロ-N-アセチルノイラミン酸(20)誘導体も多く生物界に発見されている。主なものを表-12にまとめた。生体内では各部位の体液中にあって、ウイルスに対して、活性化または不活性化作用を示すと報告されている。
| 生物名 | ヒト | ウシ | ウマ | ブタ | ラット | ニワトリ | ヒトデ |
| R(5位) | Ac | Ac Ac | Ac Gc | Ac Gc | Ac Gc Ac | Ac | Gc |
| 8位 | Me | ||||||
| 9位 | Ac | Lt Lt | Ac | ||||
| 起 源 | 尿、唾液、精液 | 尿、顎下腺 | 尿 | 尿、顎下腺 | 尿 | 血液 | 全体 |
Ac:アセチル Gc:グリコリル Lt:ラクチル Me:メチル
ヒトでは2,3-デヒドロ-N-アセチルノイラミン酸(20)が尿、唾液、精液などに発見され、牛では9位の水酸基もアセチル化された構造である。ウマではN-グリコリル誘導体が見いだされ、ブタでは9位がラクチル化されている。22)ヒトデの全身からはN-グリコリル-8-メチル誘導体が発見されるなど、面白い報告が相次いでいるから、将来、この分野から新薬が開発される可能性がある。
6 N-アセチル-2,7-アンヒドロノイラミン酸
軟らかいタイプの耳垢から、新しいシアル酸が発見されたのも、大分古い話になった。23)α型のN-アセチルノイラミン酸(22)の環が反転して、2位と7位の水酸基から分子内脱水して生成したものと考えられる。リーらは糖蛋白質を分解して、N-アセチル-2,7-アンヒドロノイラミン酸(21)に変える酵素をヒルから分離している。24)
しかし、耳垢からは微量しか得られなかったので、合成して比較確認を行なった。図-14に示すように、メチルエステルを原料として保護基をつけてから、S,S'-ビス(1-フェニル-1H-テトラゾール-5-イル)ジチオカルボナート(30)25)を反応させてS-グリコシドとして、26)分子内グリコシル化反応を行なって、45%の収率で合成した。27)
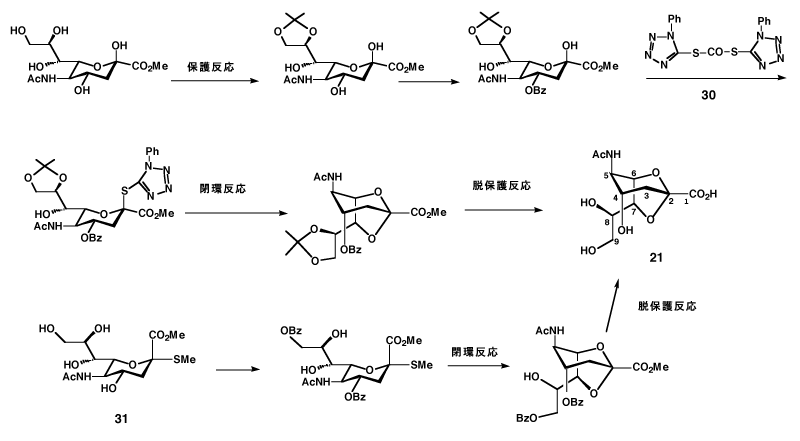
S,S'-ビス(1-フェニル-1H-テトラゾール-5-イル)ジチオカルボナート(30)は、我々の開発した強力なペプチド合成試薬である。25)分子内に窒素原子を4個ももった芳香環と、ソフトな硫黄原子をもつために各種の反応がスムースに進むのである。しかし、この方法では無理な環の反転を要求するので、収率がよくない。長谷川らの方法を用いてS-グリコシド(31)を合成し、28)これを原料とすると、収率よく目的とする化合物(21)を合成することが出来る。29)(図-13)
文献
- 1) 山川民夫、“糖脂質物語”講談社、東京、1981年; R. Schauer Ed., "Silalic Acids, Chemistry, Metabolism, and Function," Springer-Verlag, Wien, 1982.
- 2) E. Klenk, Hoppe-Seyler's Z. Physiol. Chem., 268, 50 (1941).
- 3) G. Blix, Hoppe-Seyler's Z. Physiol. Chem., 240, 43 (1936).
- 4) E. Klenk, Hoppe-Seyler's Z. Physiol. Chem., 273, 76 (1942).
- 5) G. Blix, Skand. Arch. Physiol., 80, 46 (1938). 現在では、その他に多くの発色試薬が知られており、過ヨー素酸/チオバルビツール酸(アミノフ試薬)定量法のほか、蛍光分析が一般的になっている。
- 6) G. K. Hirst, J. Exp. Med., 75, 49 (1942); idem, 76, 195 (1942).
- 7) A. Gottshalk, "The Chemistry and Biology of Sialic Acids and Related Sub-stances," Cambridge University Press, 1960.
- 8) G. Blix, A. Gottshalk, E. Klenk, Nature (London), 179, 1088 (1957).
- 9) H. Faillard, "Sialic Acids," Ed. by R. Schauer and T. Yamakawa, Kieler Verlag, Wissenshaft+Bildung, Kiel, 1988, p.6
- 10)小倉治夫、古畑公夫、有機合成化学協会誌、42, 536-543 (1984); 小倉治夫、薬学雑誌、 114, 277-303 (1994).
- 11)R. Schauer Ed., "Silalic Acids, Chemistry, Metabolism, and Function," Springer-Verlag, Wien, 1982.
- 12)H. Ogura, K. Furuhata, H. Saito, G. Izumi, M. Itoh, Y. Shitori, Chem. Lett., 1984, 1003.
- 13)A. Rosenberg, C.-L. Schengrnd Ed., "Biological Roles of Sialic Acid," Plenum Press, N.Y., 1976.
- 14)小倉治夫、古畑公夫、伊藤正善、志鳥善保、日特、昭61ー243094、(1986);小倉治夫、古畑公夫、伊藤正善、志鳥善保、日特、昭63ー28429、(1988).
- 15)H. Ogura, T. Kobayashi, K. Shimizu, K. Kawabe, K. Takeda, Tetrahedron Lett., 1979, 4745; H. Ogura, O. Sato, K. Takeda, ibid., 22, 4817 (1981); H. Ogura, K. Takeda, Heterocycles, 15, 467 (1981); 小倉治夫、武田収功、日本化学会誌、1981、836;K. Takeda, H. Ogura, Synthetic Commun., 12, 213 (1982); K. Takeda, Y. Akagi, A. Saiki, T. Tsukahara, H. Ogura, Tetrahedron Lett., 24, 4569 (1983); Fluka, Chemika-Biochemika Analytika, 1995/96, p. 612.
- 16)K. Furuhata, H. Ogura, The 50th Anniversary International Symposium on Organic Synthesis, 1992, p.127.
- 17)D. Nadano, M. Iwasaki, S. Endo, K. Kitjima, S. Inoue, Y. Inoue, J. Biol. Chem., 261, 11550 (1986); 鈴木康夫、安藤進、”ガングリオシド研究法 I”p.193, 学会出版センター、1995.
- 18)M. Nakamura, K. Furuhata, K. Yamazaki, H. Ogura, H. Kamiya, H. Ida, Chem. Pharm. Bull., 37, 2204-2206 (1989).
- 19)R. Shirai, H. Ogura, Tetrahedron Lett., 30, 2263-2264 (1989).
- 20)M. Nakamura, H. Takayanagi, K. Furuhata, H. Ogura, Chem. Pharm. Bull., 40, 879-885 (1992); H. Takayanagi, M. Nakamura, H. Ogura, XVIth International Carbohydrate Symposium, 1992, p. 518.
- 21)K. Kitajima, H. Kuroyanagi, S. Inoue, J. Ye, F.A. Troy, Y. Inoue, J. Biol. Chem., 269, 21415-21419 (1994); A. Kanamori, K. Kitajima, S. Inoue, Y. Inoue, Biochem. Biophys. Res. Commun., 164, 744-749 (1989); S. Fuchizawa, K. Furuhata, T. Matsuda, K. Kitajima, Biochem. Biophys. Res. Commun., 248, 505-510 (1998); 北島健、安形高志、仲田大輔、松田幹、糖質シンポジウム、p.73(1998); 淵澤聡子、古畑公夫、松田幹、北島健、糖質シンポジウム、p.121(1998).
- 22)小倉治夫、“複合糖質の化学と応用”小倉治夫監修、㈱シーエムシー、東京、1989、p.25;小倉治夫、ファインケミカル、15、47(1986);小倉治夫、化学と生物、29、248(1991).
- 23)M. Suzuki, A. Suzuki, T. Yamakawa, E. Matsunaga, J. Biochem., 97, 509 (1985).
- 24)Y. -T. Li, H. Nakagawa, S. A. Ross, G. C. Hansson, S. -C. Li, J. Biol. Chem., 265, 21629 (1990).
- 25)K. Takeda, K. Tsuboyama, H. Takayanagi, H. Ogura, Synthesis, 560-562 (1987); K. Takeda, K. Tsuboyama, K. Torii, M. Murata, H. Ogura, Tetrahedron Lett., 29, 4105-4108 (1988); K. Takeda, K. Tsuboyama, H. Takayanagi, R. Shirokami, M. Takeura, H. Ogura, Chem. Pharm. Bull., 37, 2334-2337 (1989).
- 26)K. Tsuboyama, K. Takeda, K. Torii, M. Ebihara, J. Shimizu, A. Suzuki, N. Sato, K. Furuhata, H. Ogura, Chem. Pharm. Bull., 38, 636-638 (1990); K. Takeda, K. Tsuboyama, K. Torii, K. Furuhata, S. Sato, H. Ogura, Carbohydr. Res., 203, 57-63 (1990).
- 27)K. Furuhata, K. Takeda, H. Ogura, Chem. Pharm. Bull., 39, 817-819 (1991).
- 28)A. Hasegawa, M. Ogawa, H. Ishida, M. Kiso, J. Carbohydr. Chem., 9, 393 (1990).
- 29)K. Furuhata, H. Ogura, Chem. Pharm. Bull., 40, 3197-3200 (1992).